الکتروشیمی و حسگرهای شیمیایی (electrochemistry and chemical sensors)-بخش اول
فصل 11
الکتروشیمی و حسگر های شیمیایی (electrochemistry and chemical sensors)- بخش اول
حسگرهای الکتروشیمیایی و حسگرهای نوری (و بیوسنسورهای مربوط ) بطور گسترده ای در سیستم های انالیزی بالینی بنیان گذاشته شده اند.سنسورهایی برای اندازه گیری گازهای خون ،الکترولیتها و متابولیتها به طور ویژه ای در دستگاه های خودکار و یا روشهای POC (point of care) و انالایزرهای موارد بحرانی بکار گرفته شده اند و این بخاطر کاربری اسان ،نگه داری اسان و توانایی اندازه گیری انالیتهای مهم بالینی در خون رقیق نشده است.هنگامی که در سیستم های کروماتوگرافی درج شوند ،اشکارسازهای الکتروشیمیایی ابزارهای بسیار حساس و انتخابی ای را برای شناسایی شماری از انالیتها مانند داروهای درمانی ،نوروترانسمیترها ،گلوتاتیون و هموسیستئین فراهم کرده است.همچنین اشکارسازی الکتروشیمیایی بطور موفقیت امیزی در پایش واکنش های انعقادی، شناسایی سرب سمی در نمونه های خون و طراحی ایمونواسی های نوین بسیار حساس بکار گرفته شده است.هنگامی که عتاصر زیستی در الکترودها درج میگردند بیوسنسورهای به دست امده توانایی انالیزی چنین دستگاه هایی را بیشتر گسترده میکنند.افزون بر این طراحی و بکار گیری اپتود (optode) که بر پایه برخی شیمی انتخابی بکار رفته در ابزارهای الکتروشیمیایی است ابزار انالیزی دیگری برای اندازه گیری گازهای خونی و الکترولیتها فراهم اورده است.
در این فصل اصول الکتروشیمیایی پایه ای 1) پتانسیومتری (potentiometry) 2) ولتامتری / امپرومتری (voltammetry/amperometry) 3) هدایت (conductance) 4) کولومتری (coulometry) خلاصه سازی شده و کاربردهای انها گفته شده است.سپس اپتودها و بیوسنسورها بحث خواهند شد.فصل شامل بحث در باره سنسورهای in vivo و کمی مهاجم هم خواهد بود.
پتانسیومتری و الکترودهای گزینش گر بون(potentiometry and ion-selective electrodes)
پتانسیومتری بطور گسترده ای در اندازه گیری بالینی pH,pCO2 و الکترولیت ها (Na+ , K+ , Cl- , Ca2+ , Mg2+ , Li+) در خون کامل،پلاسما ،سرم و ادرار و به عنوان پایه ای برای برخی بیوسنسورها در اندازه گیری متابولیتهای مورد علاقه بکار رفته است.
مفاهیم پایه (basic concepts)
پتانسیومتری اندازه گیری اختلاف پتانسیل الکتریکی بین دو الکترود (نیم -سلول ) در یک سلول الکتروشیمیایی است (شکل 11-1) به هنگامی که جریان در سلول صفر است (سلول گالوانیک ).چنین سلولی تشکیل شده است از دو الکترود (رسانای الکترون و فلزی ) که به وسیله یک محلول الکترولیت (رسانای یون ) به هم مرتبط شده اند.یک الکترود یا نیم سلول تشکیل شده است است از یک رسانای فلزی که در تماس با یک محلول الکترولیت است.رساناهای یون از یک یا چند فاز که یا در تماس مستقیم با یکدیگر هستندو یا به وسیله غشاهایی جدا شده اند که نفوذپذیر به کاتیون ها و انیون های خاص هستند.(شکل 11-1).

یکی از محلول های الکترولیت نامعلوم (unknown)و محلول ازمایش (test solution) می باشد این محلول ممکن است با یک محلول رفرانس مناسب به منظور کالیبراسیون جایگزین شود.برای اسان تر شدن سلول به گونه ای نمایش داده شده است که الکترود چپ (M L) الکترود رفرانس است و الکترود راست (M R) الکترود اندیکاتور (اندازه گیری کننده ) می باشد.
نیروی الکتروموتیو (E یا EMF) عبارت است از بیشینه اختلاف در پتانسیل بین دو الکترود (راست منهای چپ) در هنگامی که جریان سلول صفر است.پتانسیل سلول با استفاده از یک پتانسیومتر سنجیده میشود که pH متر معمولی یک نوع خاص از آن است.پتانسیومتر در واقع یک ولت متر است که پتانسیل را در سرتاسر سلول اندازه میگیرد (بین دو الکترود)، با این حال برای به دست اوردن اندازه صحیح پتانسیل که هیچ جریانی در سلول وجود نداشته باشد.این کار با کار گذاشتن یک مقاومت بالا درون ولت متر (مقاومت درونی بیش از 10^12 Ω) انجام میگردد.
درون هر یک از فازهای رسانا پتانسیل تا هنگامی که جریان صفر است ثابت است.با این حال اختلاف پتانسیل هنگامی برمیخیزد که دو فاز مختلف فازهای مختلف در تماس با یکدیگر قرار میگیرند.پتانسیل سرتاسری یک سلول الکتروشیمیایی برابر است با جمع همه شیب های پتانسیل که بین فازهای سلول وجود دارد.پتانسیل یک الکترود منفرد در رابطه با الکترولیت دربرگیرنده آن و بزرگی مطلق شیب های پتانسیل منفرد بین فازها معلوم نبوده و نمی توان اندازه گرفت .تنها اختلاف پتانسیل بین دو الکترود (نیم سلول) را متوان اندازه گرفت.شیب های پتانسیل را میتوان به صورت های 1) پتانسیل های ریدوکس (redox potentials) 2) پتانسیل های غشا (membrane potentials) 3) پتانسیل های انتشار (diffusion potentials) دسته بندی کرد.بطور کلی امکان پذیر است که سلول را طوری ساخت که همه شیبهای پتانسیل بجز یکی ثابت باشند.انگاه این پتانسیل میتواند به فعالیت یک یون خاص مورد نظر (مانند H+ , Na+) مرتبط باشد.
انواع الکترود ها (type of electrodes)
انواع گوناگونی از الکترودها در کاربری پتانسیومتری بکار گرفته میشوند.این ها شامل الکترودهای redox ، ion-selective membrane( از جنس شیشه یا پلیمر) ، و PCO2 می باشد.
الکترودهای Redox
پتانسیل های redox نتیجه تعادل شیمیایی شامل واکنشهای انتقال الکترون می باشد.
![]()
طوری که n نشاندهنده شمار الکترون های درگیر در واکنش است.هر ماده ای که الکترون ها را بپذیرد یک اکسیدان (OX) بوده و هر ماده ای الکترون بدهد یک کاهنده (Red) به شمار میرود.این دو شکل ، OX و Red ، نمایانگر یک جفت redox هستند (جفت کنژوگه redox) .معمولا فرایند های redox هموژن تنها بین دو جفت redox رخ میدهد.درچنین مواردی الکترون ها از یک کاهنده (Red1) به یک اکسیدان (OX2) منتقل میشوند.در این فرایند Red1 کنژوگه خود یعنی Ox1 اکسیده میشود در حالی که Ox2 به Red2 کاهیده میشود.
![]()
در یک سلول الکتروشیمیایی الکترون ها ممکن است از یک و یا به یک رسانای فلزی خنثی(مانند پلاتینوم ) گرفته و یا داده شود .در فرایند کاهش الکترود ها بار مثبت پیدا میکنند( الکترون ها از الکترود برداشته میشوند) و در فرایند اکسیداسیون الکترودها بار منفی می یابند (الکترون ها به الکترود افزوده میشوند).برای اسان شدن، تعادل redox ناهمگون ( معادله 2) به وسیله سلول زیر نمایش داده میشود.
![]()
یک پتانسیل مثبت (E>0) برای این سلول به این معنی است که واکنش سلول خودبخود از چپ به راست پیش میرود. E<0 به این معنی است که واکنش از راست به چپ پیش میرود و E=0 به این معنی است که دو جفت redox در تعادل دو طرفه هستند.
پتانسیل الکترود (پتانسیل کاهش =reduction potential) برای یک جفت redox عبارت است از پتانسیل جفت در رابطه با الکترود استاندارد هیدروژن که روی صفر تنظیم است (الکترود هیدروژن بحث خواهد شد ) سنجیده میشود.این پتانسیل برای اسان تر شدن موضوع به عنوان نیروی الکتوموتیو سلول در نظر گرفته میشود ، در سلول الکترود هیدروژن استاندارد به عنوان الکترود رفرانس (الکترود چپ ) و نیم سلول فرضی ،الکترود اندیکاتور (الکترود راست ) می باشد. پتانسیل کاهش (reduction potential) برای یک جفت redox مفروض به وسیله معادله نرنست (Nernst) نمایش داده میشود.

که در آن

الکترودهای redox که اکنون استفاده میشوند شامل 1) الکترودهای فلزی خنثی که غوطه ور در محلولهای دارای جفت redox هستند 2) الکترودهای فلزی که عملکرد فلزی آنها به عنوان عضوی از جفت redox است.
الکترود های فلزی خنثی (inert metal electrodes)
پلاتینوم (platinum) و طلا (gold) نمونه هایی از فلزهای خنثی هستند که برای ثبت پتانسیل redox یک جفت redox که در یک محلول الکترولیت حل شده اند بکار میروند.الکترود هیدروژن یک الکترود redox خاص است که برای اندازه گیری pH بکار میرود.این نوع ،از الکترود پلاتینوم یا طلا ساخته شده اند که به وسیله پلاتینوم بسیار پر روزنه بصورت الکترولیزی پوشانده شده است (پلاتینیزه شده است ) تا واکنش الکترود را کاتالیز نماید.


هنگامی که فشار جزیی هیدروژن (PH2) در محلول (و به این ترتیب f H2) به وسیله تشکیل حباب هیدروژن ثابت باقی ماند، انگاه پتانسیل بک تابع خطی از Log aH+ (= -pH) خواهد بود.در الکترود هیدروژن استاندارد (SHE) ، الکترود تشکیل شده است از یک محلول ابی هیدروژن کلراید با aHCl برابر با 1.000 ( یا CHCl = 1.2 mol/L) که در نعادل با فاز گازی است و با f H2 برابر با 1.000 ( یا PH2 = 101.3 kPa = 1 atm) . SHE به عنوان الکترود رفرانس نیز استفاده میشود.
الکترودهای فلزی که در واکنش های redox شرکت میکنند
الکترود نقره – کلرید نقره یک نمونه از الکترود فلزی است که به عنوان یک عضوی از جفت redox شرکت میکند.الکترود نقره –کلرید نقره از یک سیم یا میله نقره ای که با کلرید نقره (AgCl) پوشانده شده است و در یک محلول کلرید با فعالیت ثابت غوطه ور گردیده است ساخته شده است.این مجموعه یک پتانسیل نیم سلول را تشکیل میدهد.خود الکترود Ag / AgCl به عنوان یک الکترود پتانسیومتری به حساب میاید ، طوری که پتانسیل مرزی فازی آن به وسیله واکنش تعادلی انتقال الکترون ناشی از اکسایش کاهش که در سطح نقره رخ میدهد کنترل میشود.
![]()
معادله نرنست Nernst برای پتانسیل نیم سلول رفرانس برای الکترود رفرانس Ag/AgCl بصورت زیر نوشته میشود.

از انجایی که AgCl و Ag هر دو جامد هستند فعالیت انها برابر با واحد در نظر گرفته میشود ( a AgCl = a 0 Ag = 1) .بنابراین از معادله (9) بر میاید که پتانسیل نیم سلول به وسیله فعالیت یون کلراید در محلول (a Cl-) که در تماس با الکترود است کنترل میشود.
الکترود Ag/AgCl هم به عنوان یک عنصر رفرانس درونی در الکترود های پتانسیومتری گزینش یون (ISE ) بکار میرود و هم به عنوان یک الکترود نیم سلول رفرانس بیرونی با پتانسیل ثابت که برای تکمیل یک سلول پتانسیومتری لازم است. ( شکل 11-1 را ببینید).در هردو مورد الکترود Ag/AgCl باید در تعادل با یک محلول یون کلراید با فعالیت ثابت باشد.
عنصر Ag/AgCl الکترود نیم سلول رفرانس بیرونی در تماس با یک محلول بسیار غلیظ از نمک کلراید حل شونده میباشد. پتاسیم کلراید اشباع معمولا در این مورد بکار میرود.یک غشا (membrane) پر روزنه و یا پیش ماده شیشه (frit) پر روزنه برای جداسازی KCl غلیظ از محلول نمونه (sample solution) بکار میرود. فریت (frit) هم به عنوان یک سد مکانیکی برای نگه داری الکترولیت غلیظ درون الکترود بکار میرود و هم به عنوان یک سد انتشاری برای پیشگیری از انتشار پروتیین ها و دیگر گونه های موجود در نمونه به درون و قرار گرفتن در تماس با عنصر Ag/AgCl درونی.که میتواند پتانسیل انرا تخریب یا تغییر دهد. تداخل بین دو الکترولیت ناهمسان ( KCl غلیظ / کالیبراتور یا نمونه ) درون فریت رخ میدهد و یک پتانسیل تماسی مایع – مایع (Ej) شکل میگرد که منبع خطا در اندازه گیری پتانسیومتری میباشد.اختلاف پتانسیل اتصال مایع – مایع بین کالیبراتور و نمونه (پتانسیل اتصال مایع باقی مانده = residual liquid junction potential) مسوول این خطا بوده و میتوان آنرا به حداقل رساند و یا معمولا در عمل انرا در نظر نگرفت تنها اگر محتوای یونی و قدرت یونی محلول های کالیبر کننده تا جایی که ممکن است نزدیک به نمونه باشد.یک الکترولیت انتقال دهنده هم ارز (equitransferant electrolyte) در غلظتهای بالا به عنوان الکترولیت رفرانس در کم کردن پتانسیل اتصال مایع باقی مانده بیشتر کمک میکند.پتاسیم کلراید در یک غلظت برابر با ویا بیشتر از 2 مول در لیتر برای این کار بهتر است.تفاوتی نزدیک به 2% در اندازه گیری سدیم به وسیله ISE نشان داده شده است آنهم در هنگامی که غلظت KCl در الکترولیت رفرانس از 3 به 0.5 مول در لیتر کاهش یابد.بزرگی پتانسیل اتصال مایع باقی مانده را میتوان به وسیله معادله هندرسون و دانش کافی درباره دما و فعالیت یونی ،بارهای یونی و حرکت یونی برای هر الکترولیت در هر دو طرف اتصال براورد کرد.با استفاده از این براورد میتوان در مورد پتانسیل سرتاسری سلول یک اصلاح انجام داد.
وجود اریتروسیتها در نمونه ممکن است بر بزرگی پتانسیل اتصال مایع باقی مانده اثر بگذارد. برای مثال اریتروسیتها در یک خون با هماتوکریت نرمال براورد میشود که تقریبا 1.8 mmol/L خطای مثبت در اندازه گیری سدیم به وسیله ISE ایجاد کند هنگامی که یک اتصال مایع – مایع باز و نامحدود استفاده شود.این سوگرایی را میتوان با بکارگیری یک غشا یا یک فریت محدود کننده برای اصلاح اتصال مایع – مایع به کمترین اندازه رسانید.
الکترود کالومل (calomel) تشکیل شده از جیوه ای که به وسیله یک لایه از کالومل (Hg2Cl2) پوشانده شده و در تماس با یک محلول الکترولیت دارای یون کلراید (Cl-) است.الکترود کالومل به عنوان الکترود رفرانس در اندازه گیری pH به فراوانی استفاده میشود.
الکترودهای گزینشگر یون (ion-selective electrodes)
پتانسیل های غشایی با نفوذپذیری انواع خاصی از غشاها در برابر انیون ها و کاتیون های گزینش شونده ایجاد میگردد.چنین غشاهایی برای ساخت ISE استفاده میشوند که بطور گزینشی با یک گونه یونی یکتا برهم کنش میدهد.پتانسیل ایجاد شده محل رویارویی غشا – مایع نمونه متناسب با لگاریتم فعایت یونی یا غلظت یون مورد جستجو است.اندازه گیری با ISE ها ساده ،سریع و نامخرب بوده و در بازه گسترده ای از غلظتها کاربرد دارد.
غشا گزینشگر یون قلب یک ISE است چرا که در واقع گزینشگری الکترود را کنترل میکند.غشاهای گزینشگر یون از شیشه ، کریستالین و یا مواد پلیمری ساخته شده اند. محتویات شیمیایی غشا طوری ساخته شده اند تا به یک گزینشگری-نفوذپذیری مناسب به یون مورد جستجو رسید.در عمل دیگر یونها برهم کنش محدودی با جایگاه های غشایی نشان میدهند و بنابراین تا اندازه ای در اندازه گیری یک انالیت یونی تداخل ایجاد میکنند.در کاربری های بالینی اگر تداخل از مقدار مورد پذیرش بگذرد،اصلاح مقدار خوانده شده لازم است.
معادله نیکولسکی – ایزنمن (nicolsky – eisenman equation) گزینشگری یک ISE را برای یون مورد نظر در برابر یونهای مداخله گر توصیف میکند.


شیوه های گوناگونی برای تعیین گزینشگری یک ISE برای یون اصلی (primary ion) در برابر یونهای مداخله گر (interfering ion) بکار گرفته میشود.یک روش ساده ،روش محلول جداگانه (separate solution method) است که در آن پتانسیل یک ISE در محلول یونهای اصلی و محلول یونهای مداخله گر جداگانه اما با فعالیت یونی یکسان اندازه گیری میشود.ضریب گزینشگری (selectivity coefficient) انگاه از رابطه زیر محاسبه میشود.

بیشتر ISE های بکار گرفته شده در کاربردهای بالینی گزینشگری کافی را دارا هستند و نیاز به اصلاح برای یونهای مداخله گر ندارند.Oesch و همکاران ضریبهای گزینشگری برای ISE را برای یونهایی که بطور معمول در شیمی بالینی اندازه گیری میشوند را در برابر یونهایی که در خون یافت میشوند منتشر کردند.جدول 11-1 ضریبهای گزینشگری مورد نیاز را برای اندازه گیری کاتیونهای مورد نظر در شیمی بالینی در برابر کاتیونهای توانمند برای مداخله را البته با در نظر داشت بیشینه تداخل قابل پذیرش 1% برای یون اصلی ،نشان میدهد.

الکترودهای غشا شیشه ای (glass membrane electrode) و غشا پلیمری (polymer membrane electrode) دو نوع ISE هستند که در کاربردهای شیمی بالینی بطور معمول بکار گرفته میشوند.
الکترود شیشه ای (the glass electrode)
الکترودهای غشا شیشه ای برای اندازه گیری pH و Na+ و به عنوان ترانسدیوسر (transducer) درونی در حسگر های pCO2 بکار میروند.پاسخ غشاهای شیشه ای نازک به H+ اول بار در سال 1906 توسط Cremer بیان شد. در دهه 1930 کاربرد عملی این پدیده در اندازه گیری اسیدیته اب لیمو با نواوری pH متر توسط Arnold Beckman ممکن شد.الکترودهای شیشه ای از مذاب سیلیکون (silicon) و یا اکسید الومینیوم (aluminium oxide) مخلوط با اکسیدهای خاک قلیایی یا کاتیونهای فلزهای قلیایی ساخته میشوند.با تغییر در محتوای شیشه ،الکترودهایی برای گزینش H+, Na+,K+,Li+,Rb+,Cs+,Ag+,Tl+,NH4+ ساخته شده است.با این حال الکترودهای شیشه ای برای H+ و Na+ امروزه تنها الکترودهایی هستند که در برابر یونهای مداخله گر دیگر گزینش گری کافی داشته و در شیمی بالینی کاربرد پیدا کرده اند.یک فرمول معمولی برای شیشه گزینش گر H+ شامل 72% SiO2 , 22% Na2O , 6% CaO که ترتیب گزینشگری آن H+ >>> Na+ > K+ است.این غشا شیشه ای گزینشگری کافی برای H+ در برابر Na+ تا اندازه ای دارد که میتوان اندازه گیری pH را در گستره 7.0 – 8.0 در حضور Na+ >0.1 mol/L بدون خطا انجام داد.الکترودهای pH شیشه ای با ضریب گزینش گری (KH/Na) , 10^-7 در برابر Na+ و حتی بهتر از آن به دست امده است.با تغییر کوچک در فرمول غشا شیشه ای به 71% SiO2 , 11% N2O , 18% Al2O3 ترتیب گزینشگری آن H+ > Na+ > K+ میشود.بنابراین گزینشگری غشا شیشه ای برای H+ در برابر Na+ بسیار کاهش می یابد بنابراین یک حسگر عملی برای Na+ در مقدارهای pH خواهد بود که بطور معمول در خون یافت میشود.
الکترودهای غشا پلیمری (polymer membrane electrodes)
ISE های غشا پلیمری برای پایش pH و نیز برای اندازه گیری الکترولیتها شامل K+ , Na+ , Cl- , Ca2+ , Li+ , Mg2+ و CO3 (برای اندازه گیری کل CO2) بکارگرفته شده است.این نوع الکترودها غالب ترین دسته الکترودهای پتانسیومتری هستند که در دستگاه های انالیز بالینی مدرن به کار رفته اند.
مکانیسم پاسخ دهی این ISE ها در سه دسته قرار میگیرند 1) مبادله گر یون جداشده باردار (cgaged,dissociated ion exchanger) 2) ناقل همراه باردار (charged associated carrier) و 3) ناقل یون خنثی (neutral ion carrier (ionophore) ) .اولین ISE از نوع charged associated ion exchanger برای Ca2+ برای کاربردهای بالینی در دهه طراحی و تجاری گردید. این نوع ISE بر پایه ویژگی های گزینش، تعویض یون و تشکیل کمپلکس با Ca2+ ، 2-ethylhexyl phosphoric acid که در dioctyl phenyl phosphonate حل شده است طراحی شد.یک غشا پر روزنه به وسیله این مخلوط پر میشود و در انتهای بدنه الکترود چسبانده میشود.این نوع حسگر را ISE " غشا مایع (liquid membrane)) مینامند. روش دیگری طراحی شده است که به وسیله آن این ترکیب ها به درون یک غشا از جنس PVC ( پلی وینیل کلراید) بصورت پلاستیک درامده منتقل میشوند که این نوع غشا ها هم سخت و هم کاربری اسان تری دارند.این روش هم اکنون برای ساخت ISE های بر پایه PVC برای کاربردهای بالینی استفاده میشود.
یک نواوری شگرف در طراحی و کاربری معمول ISE های نوع PVC کشفی بود که به وسیله simon و همکاران انجام شد که در آن والینومایسین (valinomycin) انتی بیوتیک خنثی درون غشاهای مایع آلی (و بعدها غشاهای PVC پلاستیکی) درج میگردید و منجر به ساخت حسگری میشد که گزینشگری بالایی برای K+ در برابر Na+ دارد (K k/Na = 2.5 * 10^-4) .ISE پتاسیم بر پایه والینومایسین اولین نمونه از یک ISE ناقل خنثی (neutral carrier ISE) بود و بطور گسترده ای امروزه برای اندازه گیری معمول K+ در خون استفاده میشود.شکل 11-2 پاسخ ISE پتاسیم بر پایه والینومایسین در حضور غلظتهای فیزیولوژیک Na+ ، Ca2+ و Mg2+ را نشان میدهد.

بازه خطی گسترده این ISE تا سه برابر آن،برای اندازه گیری K+ در خون و ادرار آنرا مناسب ساخته است.بازه K+ در خون تنها یک بخش کوچکی از بازه خطی الکترود است و ΔEMF آن نزدیک به 9 mV است.تداخل دیگر کاتیون ها که بصورت سوگرایی از خطی بودن دیده شده است در فعالیتهای K+ > 10^-4 mol/L آشکار نیست.دیگر اینکه ISE های بر پایه پلیمر که توانایی گزینشگری کمی دارند ( به عنوان نمونه در اندازه گیری Mg2+ و Li+) در معرض تداخل با Ca2+ / Na+ و Na+ به ترتیب می باشند و به این دلیل نیازمند تعیین همزمان و اصلاح حضور غلظتهای چشمگیر یونهای مداخله گر است.
مطالعه ها روی ارتباط میان ساختار مولکولی و گزینشگری یونی منجر به طراحی ISE های پایه پلیمری با استفاده از شماری از یونوفور (ionophore) های طبیعی و سنتزی شده است که گزینشگری کافی برای کاربردهای انالیز بالینی دارند.ساختار شیمیایی چندتا از این یونوفورهای خنثی در شکل 11-3 به نمایش گذاشته شده است.

الکترودهای بر پایه مبادله انیون جدا شده (dissociated anion exchanger-based electrodes) از نمکهای امونیوم چهارظرفیتی چربی دوست (lipophilic quaternary ammonium salts) به عنوان احزای فعال غشایی برای تعیین یون Cl- در خون کامل ،سرم ،و پلاسما با وحود محدودیت هایی هنوز بصورت تجاری استفاده میکند. گزینشگری این نوع ISE به وسیله استخراج یون به درون فاز آلی غشا کنترل میشود و تابعی است از ویژگی چربی دوستی یون ( چون برخلاف حامل های پیشتر توصیف شده ، هیچ برهم کنش اتصالی مستقیمی بین جایگاه مبادله گر و انیون در فاز غشایی رخ نمیدهد).بنابراین ترتیب گزینشگری برای ISE اندازه گیری کننده Cl- بر پایه مبادله گری انیون به صورتR- > ClO4- > I- > NO3- > Br- > Cl- > F- . بنابراین بکارگیری الکترود مبادله گر یون Cl- محدود به نمونه هایی است که غلظتهای چشمگیری از انیون های چربی دوست تر از Cl- در آنها نباشد.برای مثال نمونه های خونی که سالیسیلات یا تیوسیانات در انها وجود دارد در اندازه گیری Cl- تداخل مثبت ایجاد میکند.چنانچه الکترود در معرض مکرر ضد انعقاد هپارین قرار گیرد منجر به از دست رفتن حساسیت الکترود نسبت به Cl- میشود چون هپارین با بارهای منفی به درون غشا استخراج شده است.در واقع این فرایند استخراج بطور موققیت امیزی در طراحی روشی برای تشخیص غلظتهای هپارین در خون به روش پتانسیومتری بکار گرفته شده است، همچنین در طراحی روشهای پتانسیومتری ساده برای غربالگری وجود الاینده های پلی انیونی سمی و با چگالی بالای بار الکتریکی (مانند کندروئیتین سولفات بسیار سولفاته ) در فراورده های هپارین با گرید (grade) زیست پزشکی استفاده شده است.
گزینشگری بالا برای انیون کربنات با یونوفور حامل خنثی (neutral carrier ionophore) که دارای گروه های تری فلوئورو استوفنون (trifluoroacetophenone) است و به درون یک غشا پلیمری پوشانیده میشود حاصل میگردد. حاصل افزوده شدن یونهای کربنات به چنین یونوفورهایی ،یونوفورهایی با بار منفی است و الکترودهای حاصل در دستگاه های تجاری شده برای تعیین کل دی اکسید کربن در سرم یا پلاسما پس از رقیق سازی خون به مقدار pH بین 8.5 تا 9.0 ، بازه ای که بخش چشمگیری از کل دی اکسید کربن به شکل انیون های کربنات وجود دارد ، بکار گرفته میشود.
یک فرمول معمولی از ISE های با غشا PVC که در تجهیزهای بالینی بکار برده میشود در زیر اورده شده است.

پلاستی سایزر (plasticizer) در کنترل میزان قطبی بودن غشا نقش مهمی دارد و بنابراین در کنار یونوفور یک نقش مهمی در تعیین میزان گزینشگری غشا در مورد یون جستجو شونده بازی میکند.یک انیون لیپوفیل بزرگ ( مانند مشتق تترافنیل بورات )اغلب به عنوان یک افزودنی در ساخت غشا های ISE گزیشنگر کاتیون بکار گرفته میشود.این انیون به عنوان یک انیون مقابل برای کاتیون جستجو شونده عمل میکند، این کاتیون ها به درون فاز غشا استخراج میشوند و با یونوفور خنثی کمپلکس هایی با بار مثبت تشکیل میدهند.با این حال نسبت مکان های یونوفور متصل به نامتصل در سطح غشا است که بزرگی پتانسیل مرزی فازی (phase boundary potential) تولید شده در غشا ISE را تعیین میکند.بنابراین پاسخ گزینشی به فعالیت یون جستجو شونده یک ویژگی برهم کنشی برای یک غشا ISE فرضی به شمار میرود.
بررسی ها نشان داده است که حد تشخیص نهایی در ISE های غشا پلیمری (polymer membrane-type ISE) تا حدی به وسیله نشت یونهای انالیت از محلول درونی (internal solution) به سطح بیرونی غشا و به درون فاز نمونه که در تماس نزدیک با غشا است کنترل میگردد.به این ترتیب کمترین حد تشخیص به وسیله کاهش در غلظت یون انالیت اصلی که در محلول درونی (محلول اینترنال ) الکترود است ، به دست می اید.دیگر اینکه این نشت یونهای انالیت همراه با یک فرایند تعویض – یون در محل رویارویی نمونه و غشا ،هنگامی که انتخاب گری غشا در برابر دیگر یونها در دست بررسی است ، اغلب یک ضریب انتخاب گری پتانسیومتری را به دست میدهد که براوردی است از میزان انتخاب گری واقعی غشا.برای تعیین ضریب انتخابگری منحرف نشده (unbiased selectivity coefficients) به روش محلول جدا ، می بایست غشا برای مدت زمان زیادی در معرض یونهای انالیت قرار نگیرد و دیگر اینکه غلظت یونهای انالیت در محلول اینترنال پایین باشد.برای پیشگیری از نشت یونهای اصلی از محلول درونی ISE های غشا پلیمری معمول ، طراحی های بسیار پایدار نوینی برای سنسورهای یونی حالت جامد پیشنهاد شده است که در آن غشا حسگر یون بر پایه پلیمر درج شده به وسیله یونوفور ( بر پایه پلی (متیل متاکریلات) / پلی (دسیل متاکریلات کو پلیمر) روی یک لایه پلیمری رسانا از جنس پلی (3-اکتیل اتیوفن 2،5 –داییل ) پوشش داده میشود و کل این ساختار روی سطحی از الکترود طلا قرار میگیرد.
یک کاربرد جالب الکترودهای غشا پلیمری (یا شیشه ای ) انتخاب گر سدیم در تعیین هماتوکریت کل خون است.از انجایی که غلظت سدیم درون سلولی بسیار کمتر از فاز پلاسمایی است ، تغییر در غلظت سدیم (رقیق شدن ) اندازه گیری شده به روش پتانسیومتری پیش و پس از لیز اریتروسیت ها میتواند در اندازه گیری هماتوکریت نمونه خون بکار گرفته شود.
این شیوه میتواند همراه شود با اندازه گیری همزمان تغییرها در غلظت یون پتاسیم که به وسیله ISE غشا پلیمری بر پایه والینومایسین انجام میشود و به این شیوه غلظت یونهای پتاسیم درون گلبولهای قرمز اندازه گیری شود.
الکترودها برای pCO2 (electrodes for pCO2)
الکترودهایی برای اندازه گیری pCO2 در مایع های بدن طراحی شده است.اولین الکترود pCO2 در دهه 1950 به وسیله Stow و severinghaus طراحی شد که از یک الکترود pH شیشه ای به عنوان یکی از عنصرهای درونی در یک سلول پتانسیومتری برای اندازه گیری فشار جزیی دی اکسید کربن استفاده میکرد.این پیشرفت مهم راه انالایزرهای خونی سه کاناله (pH , pCO2 , pO2) را به بازار هموار کرد تا به این وسیله تصویر کاملی از میزان اکسیژنه شدن و وضعیت اسید – باز خون به دست اید.
شکل 11-4 یک نمودار از الکترود stow-severinghaus را برای pCO2 نشان میدهد.

یک غشا نازک ( = 20 um) که تنها به گازها و بخار اب نفوذپذیر است در تماس با نمونه قرار میگیرد.غشا هایی از جنس لاستیک سیلیکونی ، تفلون و دیگر مواد پلیمری برای این منظور مناسب هستند.در سمت مخالف غشا یک لایه نازک از الکترولیت که از یک نمک ضعیف بیکربنات ( در حدود 5 mmol / L) و یک نمک کلراید تشکیل شده است، قرار دارد.یک الکترود pH و یک الکترود رفرانس Ag/AgCl در تماس با این محلول قرار دارد.گاز دی اکسید کربن از نمونه یا ماتریکس کالیبراسیون از طریق غشا انتشار می یابد و در لایه الکترولیت اینترنال حل میشود.اسید کربنیک تشکیل شده و تجزیه میگردد و pH محلول بی کربنات در لایه اینترنال طبق رابطه زیر تغییر میکند.

رابطه بین pCO2 نمونه و سیگنال تولید شده به وسیله الکترود pH اینترنال لگاریتمی بوده و از معادله نرنست پیروی میکند.الکترود را میتوان با استفاده از مخلوط گازهای دقیق و یا با استفاده از محلول هایی با غلظتهای پایدار pCO2 کالیبر کرد.اگر چه الکترودهای مدل severinghaus برای pCO2 در انالایزرهای گازهای خونی بسیار گسترش پیدا کرده است اما چنین چارچوبی
محدود است به اندازه ،شکل ،و گنجاندن بخش حساس به pH اینترنال.
یک سلول پتانسیومتری کمی متفاوت برای اندازه گیری pCO2 در شکل 11-5 نشان داده شده است.
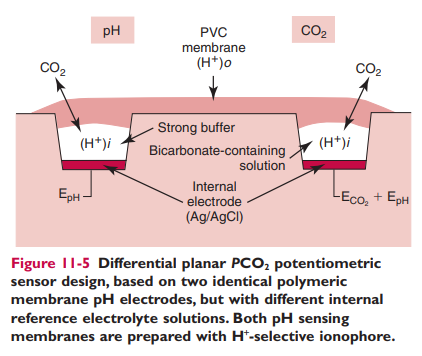
این سلول از دو الکترود گزینشگر pH از نوع PVC در دو حالت جداگانه بهره میگیرد.غشاهای الکترود دارای یک یونوفور خنثی از نوع امین و لیپوفیل هستند که یک گزینشگری بسیار بالایی برای H+ از خود نشان میدهند.(شکل 11-3 را ببینید).یک الکترود دارای یک لایه اینترنال است که بافر شده است و دیگری بافر نشده و دارای یک غلظت پایینی از نمک بی کربنات است.گاز دی اکسید کربن از نمونه و یا ماتریکس محلول کالیبراسیون در سرتاسر غشای بیرونی و از جنس PVC گزینشگر H+ در هر دو سنسور انتشار می یابد.در سمت بافر نشده (unbuffered) ، انتشار CO2 یک سوگرایی پتانسیل در محل رویارویی درونی غشا پاسخ دهنده به pH، در تناسب با غلظت pCO2 نمونه ایجاد میکند.سیگنال در الکترود با لایه درونی بافر شده، از CO2 ای که در سراسر غشا انتشار می یابد،اثر نمی پذیرد.در پایان، یک نیمه از سنسور تنها به pH پاسخ میدهد و نیمی دیگر هم به pH و هم به pCO2 پاسخ میدهد.اختلاف سیگنال بین دو الکترود هر گونه اثر pH نمونه ،در پتانسیل سلول اندازه گیری شده کلی را از بین میبرد. اختلاف سیگنال بنابراین تنها متناسب با pCO2 خواهد بود.برخلاف الکترودهای مدل Severinghaus این گونه سنسورهای pCO2 پتاسیومتری در شکل صفحه ای تجاری شده اند و سازگاری بسیار اسانی در ساخت انبوه در ردیفهای سنسوری دارند.
پتانسیومتری مستقیم به وسیله ISE
– واحدهای اندازه گیری و گزارش برای کاربردهای بالینی
روشهای انالیزی قدیمی مانند فلیم فوتومتری برای اندازه گیری الکترولیتها غلظت کلی (c) یک یون مفروض را در نمونه فراهم میاوردند که معمولا به صورت واحد میلی مول های یون در یک لیتر نمونه (mmol/L) بیان میشد.مولالیتی (m) اندازه ای است از مول های یون در جرمی از اب (mmol/Kg) در نمونه.با استفاده از یون سدیم به عنوان نمونه ،ارتباط بین غلظت و مولالیتی از رابطه زیر به دست میاید.
![]()
که pH2O غلظت جرمی اب به صورت Kg/L است.برای پلاسمای خون نرمال ،غلظت جرمی اب تقریبا برابر با 0.93 Kg/L است اما در نمونه هایی با لیپید یا پروتیین های افزایش یافته این مقدار ممکن است تا 0.8 Kg/L پایین بیاید.در این نمونه ها اختلاف بین غلظت و مولالیتی ممکن است به بزرگی 20% باشد.یک برتری چشمگیری که روش پتانسیومتری مستقیم به وسیله ISE در اندازه گیری الکترولیتها دارد این است که این تکنیک به مولالیتی حساس است و بنابراین از تغییر در غلظت پروتیین و لیپیدها در نمونه اثر نمی پذیرد.تکنیک هایی مانند فلیم فوتومتری،روشهای ISE که نیاز به رقیق سازی نمونه دارد (پتانسیومتری غیر مستقیم) و دیگر روشهای پتانسیومتری که نیاز به رقیق سازی نمونه دارند از پروتیین ها و لیپیدها تاثیر میپذیرند.در این روشها تنها فاز آبی نمونه رقیق میشود و منجر به نتایجی کمتر از مولالیتی نمونه ،به شکل تابعی از غلظت پروتیین و لیپید نمونه میگردد.بنابراین خطر رخداد خطا وجود دارد مانند غلظتهای پایین سدیم به شکل کاذب که در مواردی همچون غلظتهای بسیار افزایش یافته پروتیین و لیپیدها رخ میدهد.
افزون بر تفاوت بین مولالیتی و غلظت ، اندازه گیری یونها به وسیله پتانسیومتری مستقیم واحد دیگری از اندازه گیری را فراهم میکند به نام فعالیت (activity=a) که عبارت است از غلظت یون ازاد و نامتصل در محلول.بر خلاف روش هایی که حساس به غلظت یون هستند،ISE ها وجود یونهای پیچیده و یا به شکل الکتروستاتیکی مسدود شده را شناسایی نمیکنند.ارتباط بین فعالیت و غلظت دوباره با استفاده از یون سدیم به عنوان نمونه ، به شکل زیر بیان میشود.
![]()
که در آن ϒ یک مقدار بدون واحد است که ضریب فعالیت (activity coefficient) نام دازد.ضریب فعالیت در اصل وابسته به قدرت یونی (ionic strength) نمونه دارد که به وسیله معادله debye-huckel تعریف میگردد.

که در آن A و B ثابتهای وابسته به دما هستند ( A=0.5213 و B=3.305 در اب در دمای 37C است)، a پارامتر اندازه یونی برای یک یون خاص است و I قدرت یونی است ( I = 0.5 Σm * z^2 که در ان z شمار بار یون ها است). معادله 16 نشان میدهد که کاهش در ضریب فعالیت با افزایش در قدرت یونی رخ میدهد.این اثر هنگامی که بار (z) یونی بسیار بالا است بیشتر نمود پیدا میکند. ضریب فعالیت یونها در مایع های زیستی مانند خون و سرم را با صحت بالا به اسانی نمیتوان براورد کرد چون میزان همکاری ماکرومولکولهایی مانند پروتیین ها در قدرت یونی کلی نامعلوم است.با این حال به در نظر گرفتن اینکه قدرت یونی نرمال پلاسمای خون برابر با 0.16 mol/Kg است، براورد ضریب فعالیت در 37 C به این ترتیب است که Na+ = 0.75 , K+ = 0.74 , Ca2+ = 0.31 .با توجه به معادله 15 فعالیت و ضریب در نمونه ها با قدرت یونی فیزیولوژیک به ویژه برای یونهای دو ظرفیتی به میزان زیادی فرق میکند.
از نظر فیزیولوژی،فعالیت یونی در تعادل های شیمیایی و یا فرایند های زیستی نسبت به غلظت بسیار مرتبط تر است. با این حال در عمل غلظت یونی در ازمایش های بالینی واژه اشناتری است و پایه بازه های مرجع و غلظتهای بالینی برای تصمیم گیری در مورد الکترولیتها را تشکیل میدهد.در اغاز تکامل ISE ها به عنوان ابزارهای عملی در شیمی بالینی ،تصمیم بر این شد که تغییر بازه های مرجع بالینی به یک سیستمی بر پایه فعالیت بجای غلظت غیر عملی بوده و خطر تفسیرهای بالینی نادرست وجود دارد.یک شیوه عملی در استفاده از ISE ها در انالایزرهای نوین بدون تغییر بازه های مرجع بر پایه غلظت، ساخت محلولهای کالیبراسیونی است که قدرت یونی و محتویات یونی ان تا جای ممکن نزدیک به پلاسمای خون است. در این شیوه ضریب فعالیت هر یون در محلولهای کالیبراسیون نزدیک به آن چیزی است که در ماتریکس نمونه است و این اجازه کالیبراسیون و اندازه گیری الکترولیت ها را با واحد غلظت بجای فعالیت میدهد.
یک مجموعه معمولی از محلول ها برای کالیبراسیون چندین ISE در یک انالایزر در جدول 11-2 نشان داده شده است.

برای کالیبر کردن هر ISE دو نقطه لازم است.اختلاف در پتانسیل سلول تولید شده توسط این دو محلول (ΔE) برای اندازه گیری شیب پاسخ سلول ( slope = ΔE / Δlog c) استفاده میشود که c غلظت یون در هر یک از محلول های کالیبراسیون است و جایگیزین فعالیت شده است.پتانسیل الکترود استاندارد E0 به عنوان نقطه نقطه تقاطع با محور y ها در نظر گرفته میشود.تعیین غلظت یون در یک نمونه نامعلوم یک حل ساده معادله nockolsky-eisenman (معادله 10( است البته پس از اینکه پتانسیل سلول توسط نمونه ایجاد شده و اندازه گیری شود.
کالیبراسیون سلول با واحد غلظت انجام میشود.با این حال همانگونه که پیشتر گفته شد، پتانسیومتری مستقیم به مولالیتی یون حساس است و مولالیتی یون مرتبط است با غلظت محتویات اب در نمونه (معادله 14( .محتویات ابی محلول کالیبر کننده نشان داده شده در جدول 11-2 تقریبا برابر است با 0.99 kg / L . محتویات ابی پلاسمای خون نرمال تقریبا برابر است با 0.93 kg/L . مولالیتی 7% بزرگتر از غلظت در این پلاسمای نرمال می باشد.سلول پتانسیومتری مستقیم نتیجه ها را تقریبا 6% بزرگتر از غلظت در نمونه نرمال گزارش میکند و این بخاطر اختلاف در محتویات اب در نمونه و اب کالیبراتور (0.99 / 0.93 = 1.06) است.
پتانسیومتری مستقیم روش برتری به شمار میاید چون پروتیین ها و لیپیدهای نمونه بر این تکنیک اثر نمیگذارند، با این حال بکار بردن بازه مرجع بالینی بر پایه غلظت باعث اشتباه و تفسیر نادرست بالینی میگردد.بیشتر سازندگان سیستم های اندازه گیری الکترولیت ها بر این مشکل غلبه کرده اند، با بکار گیری راهنمایی های انستیتوی استانداردهای ازمایشگاهی و بالینی(CLSI) که استفاده از فاکتورهای همخوانی (correlation factor) را برای استاندارد کردن اندازه گیری های ISE به واحدهای غلظت توصیه میکند.این فاکتورها میتوانند به وسیله استاندارد کردن اندازه گیری های ISE با استفاده از مواد مرجع گواهی دار (certified reference materials) که پایه سرم انسانی دارند و ارزش های الکترولیتی آنها با واحد غلظت مشخص گردیده است ، بدست ایند.سپس فاکتورهای همخوانی با استفاده از یک الگوریتم که در نرم افزار دستگاه وجود دارد برای محاسبه نمونه ها بکار گرفته میشود.
ولتا متری / امپرومتری (voltammetry / amperometry)
روشهای ولتامتری و امپرومتری از حساسترین و پرکاربردترین روشهای الکتروانالیزی است.
مفاهیم پایه (basic concepts)
برخلاف روش پتانسیومتری ،روشهای ولتامتری و امپرومتری برپایه سلولهای الکترولیزی الکتروشیمیایی است طوری که در آن یک ولتاژ بیرونی به یک الکترود در حال کار قطبی شونده (که در برابر یک الکترود رفرانس مناسب سنجیده میشود : Eappl = Ework = Eref)،بکار گرفته میشود و سپس جریان کاتدی ( برای کاهش های انالیزی ) یا جریان اندی ( برای اکسایش های انالیزی ) ردیابی و اندازه گیری میشوند که متناسب است با غلظت انالیت در نمونه ازمایش.میزان جریان تنها اگر Eappl بزرگتر از یک ولتاژ معین (ولتاژ تخلیه ) باشد ، برای یک واکنش کاهش – اکسایش مفروض ( Ox + ne- < --- > Red : به وسیله مقدار E0 برای آن واکنش (پتانسیل کاهش استاندارد) تعیین میشود) به وسیله ترمودینامیک و به وسیله کینتیک انتقال الکترون ناهمگون در محل رویارویی الکترود درحال کار تعیین میشود.اغلب کینتیک اهسته انتقال الکترون برای واکنش کاهش – اکسایش روی یک الکترود درحال کار خنثی ( پلاتینوم ،کربن ،طلا و مانند اینها ) استفاده از Eappl بسیار منفی تر (برای کاهش ) یا مثبت تر (برای اکسایش ) را نسبت به آنچه که پیش بینی میشود الزامی میکند البته با توجه به E0 برای واکنش کاهش – اکسایش مفروض .به این اوورپتانسیل (overpotential=ŋ) گفته میشود.جدای از اینکه برای انتقال الکترون اورپتانسیل وجود دارد یا نه یک واکنش کاهش یا اکسایش خاص در سطح الکترود در حال کار در ولتامتری / امپرومتری رخ میدهد و این همان انتقال بار ( جریان ) است که در این محل رویارویی رخ میدهد و اطلاعات انالیزی را فراهم میکند.
برای سلولهای الکترولیزی که پایه روشهای ولتامتری و امپرومتری را شکل میدهند رابطه زیر برقرار است.
![]()
که در آن Ecell پتانسیل ترمودینامیک بین الکترودهای کار و رفرانس در غیاب یک ولتاژ بیرونی محض می باشد.هنگامی که ولتاژ بیرونی بزرگتر و یا کمتر از این پتانسیل تعادل است ، مثبت و منفی هر گونه اورپتانسیل (ŋ) به دلیل واکنش کاهش یا اکسایش در الکترود کار سبب راه اندازی جریان خواهد کرد.ولتاموگرام (voltammogram) نموداری است از جریان دیده شده ، i ، در برابر Eapp

در امپرومتری (amperometry) یک ولتاژ ثابت بکار برده میشود و جریان ایجاد شده پایش میگردد.مقدار این جریان بطور معکوس در ارتباط است با مقاومت (resistance) محلول الکترولیت و نیز در ارتباط است با هر گونه مقاومت اشکار که از انتقال جرم گونه های انالیت به سطح الکترود کار ایجاد میگردد. از آنجایی که واکنشهای الکتروشیمیایی ناهمگون هستند و تنها در سطح الکترود کار رخ میدهند ،مقدار جریان دیده شده همچنین بسیار وابسته به سطح (A) الکترود کار می باشد.
هنگامی که یک پتانسیل در الکترود کار بکار برده میشود که سبب اکسایش یا کاهش در گونه های موجود در فاز محلول در تماس با الکترود میگردد، واکنش الکتروشیمیایی سبب میشود که غلظت گونه های الکترواکتیو در سطح الکترود کاهش یابد ( شکل 11-7) ، فرایندی که قطبی شدن غلظت (concentration polarization) نامیده میشود.

این وضعیت سبب میشود که یک شیب غلظت از گونه های انالیت بین محلول نمونه عمده (bulk sample solution) و سطح الکترود ایجاد گردد. هنگامی که محلول عمده تکان داده میشود،لایه انتشار انالیت با سرعت زیاد به یک فاصله ثابت از سطح، تغییر می یابد که کنترل آن به وسیله شدت تکان دادن محلول است.این لایه انتشار ،لایه نرنست (nernst layer) نیز نامیده میشود و یک ضخامت معینی (ᵟ) دارد که این ضخامت معین پس از مدت زمان کوتاهی (شکل 11-7 را ببینید) هنگامی که محلول جابجا میشود برقرار میگردد.ولتامتری در حضور جابجایی ( با تکان دادن محلول ، چرخاندن الکترود جریان دادن محلول در کنار الکترود و همانند اینها ) انجام میشود و به آن ولتامتری حالت پایدار (steady-state voltametry) گفته میشود.هنگامی که محلول حرکت داده نمی شود، لایه انتشار با گذر زمان بیشتر و بیشتر رشد میکند ( به عبارت دیگر ثابت نخواهد بود) ،و مقدارهای ᵟ بزرگ و بزرگتری با گذر زمان ایجاد میکند.به این، ولتامتری حالت ناپایدار (non-steady-state voltametry) گفته میشود و اغلب سبب جریان های قله ای شکل در نمودار i در برابر E appl در سلولهای الکترولیزی میگردد.
در ولتامتری حالت پایدار هنگامی که پتانسیل الکترود کار اسکن میشود پس از مقداری که سبب واکنش الکتروشیمیایی میشود، جریان به سرعت افزایش می یابد و سپس هموار میگردد حتی در صورتی که E appl بیشتر تغییر می یابد. شکل 11-6 یک چنین موجی را برای یک واکنش کاهش فرضی در مورد یک گونه اکسید شده (Ox) نشان میدهد که از طریق n الکترون به یک گونه کاهش یافته (Red) کاهش می یابد.هنگامی که پتانسیل بکار رفته بسیار منفی تر از میزان مورد نیاز است، جریان به یک مقدار محدود کننده ( که جریان محدود کننده ، il نامیده میشود) میرسد.این جریان محدود کننده متناسب است با غلظت گونه های الکترواکتیو (در این مورد Ox) که طبق معادله زیر بیان میشود.
![]()
که در آن i جریان اندازه گیری شده به امپر است، n برابر است با شمار الکترون ها در واکنش الکتروشیمیایی ( در این مورد کاهش ) ،F ثابت فارادی است ( 96487 coulombs/mol) ،A سطح الکتروشیمیایی الکترود کار است ( به cm2 ، با فرض ژئومتری صفحه ای برای الکترود) ، D ضریب انتشار ( به cm2/sec) گونه های الکترواکتیو است ( در این مورد Ox) ، ᵟ ضخامت لایه انتشار است (به cm) ، و C غلظت گونه های انالیت است به mol/cm3 . عبارت D/ᵟ اغلب به صورت m0 بیان میشود که ضریب انتقال جرم گونه های Ox به سطح الکترود کار است.توجه کنید که معادله (18) نشان میدهد که یک رابطه خطی بین جریان محدود کننده و غلظت برقرار است.دقیقا همان رابطه برای تشخیص گونه های کاهش یافته به وسیله واکنش اکسایش در الکترود کار بکار برده میشود.در این مورد طبق قرارداد، جریان اندی حاصل به عنوان جریان منفی در نظر گرفته میشود. همانطور که در شکل 11-6 نشان داده شده است پتانسیلی از الکترود کار که مطابق با جریانی است که دقیقا نصف جریان محدود کننده است، E1/2 نامیده میشود.این مقدار وابسته به غلظت انالیت نیست.E1/2 به وسیله ترمودینامیک (E0) واکنش اکسایش – کاهش مفروض ، وضعیت محلول (مانند ،اگر پروتون در واکنش درگیر است انگاه pH بر مقدار E1/2 اثر میگذارد) و هر گونه اورپتانسیل ایجاد شده به وسیله انتقال اهسته الکترون در سطح الکترود کار، تعیین میگردد.مقادیر E1/2 نشانگر یک گونه مفروض است که واکنش الکتروشیمیایی را در شرایطی معین متحمل میشود و بنابراین مقادیر E1/2 تشخیص یک گونه الکترواکتیو را از گونه دیگر در یک نمونه یکسان فراهم میکند.اگر مقادیر E1/2 برای گونه های مختلف به میزان چشمگیری ( مثلا >120 mV) تفاوت داشته باشند انگاه اندازه گیری چندین جریان محدود کننده در یک ولتاموگرام مفروض نتایج کمی را برای چندین گونه مختلف به طور همزمان فراهم میکند.
سلولهای الکتروشیمیایی که برای اندازه گیری های ولتامتری و یا امپرومتری بکار گرفته میشوند میتوانند دو یا سه پیکربندی داشته باشند.در حالت دو الکترودی، ولتاژ بیرونی بین الکترود کار و الکترود رفرانس بکار میرود و جریان پایش میشود. از انجایی که جریان باید از طریق الکترود رفرانس نیز بگذرد ، یک چنین گذر جریانی میتواند غلظت گونه های الکترواکتیو در سطح الکترود را که وظیفه اماده نگه داشتن پتانسیل نیم سلول الکترود رفرانس را دارند را تغییر دهد، تغییر مقدار به وسیله فرایند قطبی شدن غلظت (concentration polarization) رخ میدهد.برای نمونه اگر یک الکترود رفرانس Ag/AgCl در یک سلول بکار رفته باشد که در ان واکنش کاهش برای انالیت در الکترود کار رخ میدهد انگاه واکنش اکسایش در سطح الکترود رفرانس رخ خواهد داد .
![]()
بنابراین فعالیت / غلظت یونهای کلراید نزدیک سطح الکترود کاهش خواهد یافت که پتانسیل الکترود رفرانس نسبت به مقدار حالت تعادل واقعی آن بر پایه فعایت واقعی یون کلراید در نیم سلول رفرانس بیشتر مثبت خواهد بود چون در معادله Nernst برای این نیم سلول داریم.
![]()
به وسیله بسیار پایین نگاه داشتن چگالی جریان (J ; amperes/cm^2) در الکترود رفرانس میتوان از چنین قطبش غلظتی در الکترود رفرانس جلوگیری کرد.در عمل میتوان با گزینش یک سطح الکترود کار بسیار کوچک در سلول الکتروشیمیایی نسبت به سطح الکترود رفرانس به این هدف دست یافت، به این ترتیب گذر جریان به وسیله این سطح بسیار کوچک محدود خواهد شد و مقدارهای J همانطور که توضیح داده شد برای رفرانس بسیار کوچک خواهد بود و از قطبش غلظتی پیشگیری میکند.
برای اینکه تغییرات به طور کامل در پتانسیل نیم سلول الکترود رفرانس از میان برداشته شود یک پیکربندی سه الکترودی اغلب بکار برده میشود.به زبان ساده در این ساختار یک ولتاژ در الکترود کار بکار برده میشود که که در برابر الکترود رفرانس اندازه گیری میشود البته به روش اندازه گیری پتانسیومتری نوع جریان صفر، اما گذر جریان بین الکترود کار و یک الکترود سوم به نام کانتر الکترود (counter electrode) برقرار است.بنابراین کاهش در الکترود کار رخ دهد ،اکسایش در کانتر الکترود رخ میدهد اما هیچ واکنش خالصی در سطح الکترود رفرانس رخ نخواهد داد چون هیچ گذر جریانی از طریق این الکترود رخ نمیدهد.
در روشهای ولتامتری ،Eappl تغییر موجی شکل دارد و سبب تغییر پتانسیل الکترود کار بصورت تابعی از زمان شده و جریان ایجاد شده اندازه گیری میشود.تغییر جریان در گستره پتانسیل تخلیه رخ میدهد که هنگامی که برای یک انالیت اختصاصی باشد این تغییر جریان در بهترین حالت خواهد بود.با این حال مکان پاسخ جریان بصورت تابعی از Eappl داده هایی درباره ماهیت گونه های موجود ( به عنوان مثال E1/2) بهمراه سیگنال وابسته به غلظت فراهم میکند.این اسکن Eappl میتواند خطی ( linear sweep voltametry) یا میتواند شکل های پیچیده تر داشته باشد که حساسیت افزایش یافته ای را برای اندازه گیری غلظت گونه ای الکتروشیمیایی فعال فراهم میاورد (به عنوان نمونه normal pulsed voltametry, differential pulse voltametry,square wave voltametry ) .هنگامی که از DME ( dropping mercury electrode) استفاده شود چنین روشهای ولتامتری را به عنوان روشهای پلاروگرافی انالیز می شناسند.
روشهای امپرومتری با ولتامتری فرق دارند چون در روشهای امپرومتری Eappl بطور کلی در مقدار پتانسیلی ثابت میشود که ناحیه پلاتوی جریان محدود کننده در ولتاموگرام وجود دارد و سپس جریان حاصل به سادگی پایش میشود که متناسب است با غلظت. امپرومتری نسبت به روشهای ولتامتری معمول بسیار حساستر است چون جریان های شارژ کننده پس زمینه که از تغییرات Eappl به صورت تابعی از زمان ،در ولتامتری ریشه می گیرند در روش امپرومتری وجود ندارند. به این ترتیب هنگامی که انتخاب گری در یک Eappl مفروض تضمین شده باشد ، امپرومتری نسبت به روشهای ولتامتری برای اندازه گیری های کمی بسیار حساس، برنری دارد.
کاربردها (applications)
اکسیژن مولکولی توانایی پذیرش چندین واکنش شیمیایی را داراست که همه انها با یک اورپتانسیل چشمگیری در الکترودهای جامد مانند Pt , Au , Ag همراه است .برای نمونه در واکنش زیر ،

یک E1/2 در نزدیکی های -0.500 V روی الکترود Pt (در برابر یک الکترود رفرانس Ag/AgCl ) با یک پلاتویی در جریان محدود کننده که در نزدیکی های -0.600 V شروع میشود از خود نشان میدهد.این واکنش برای پایش PO2 در خون بکار میرود که پایه حسگرهای امپرومتری اکسیژن به سبک کلارک است (شکل 11-8).

این ابزار از یک الکترود صفحه ای کوچک از جنس پلاتینوم به عنوان الکترود کار ( که در یک شیشه یا ماده جداکننده دیگر بسته بندی شده است) و یک الکترود Ag/AgCl نیز به عنوان الکترود رفرانس معمولا با طرح استوانه ای بهره میگیرد (شکل 11-8 را ببینید).این سلول دو الکترودی الکترولیتی درون یک محافظ سنسور جایگذاری میشود طوری که یک ممبران نفوذپذیر به گاز ( مانند پلی پروپیلن ، لاستیک سیلیکونی ،تفلون) در پایانه دور آن تعبیه شده است.الکترود کار درونی از جنس پلاتینوم به شدت در برابر ممبران نفوذ پذیر به گاز فشرده میشود طوری که یک فیلم نازک از محلول الکترولیت درونی (معمولا بافر به همراه KCl افزوده شده ) ایجاد گردد.اکسیژن نمونه میتواند از سرتاسر ممبران نفوذ کند و طبق واکنش الکتروشیمیایی بالا کاهش یابد. یک E appl
-0.650 یا -0.700 ولت در برابر Ag/AgCl (در گسترده جربان محدود کننده ) به الکترودکاری از جنس پلاتینوم منجر به یک جریانی میشود که متناسب است با PO2 موجود در نمونه ( شامل خون تام ).در غیاب اکسیژن جریان در این ولتاژ بکار رفته با شرایط امپرومتری بسیار نزدیک صفر خواهد بود.
ممبران بیرونی نفوذپذیر به گاز، الکترود کلارک را قادر میسازد که اکسیژن را با گزینشگری بسیار بالا نسبت به دیگر گونه های به اسانی کاهش یافته که ممکن است در نمونه مفروض وجود داشته باشند (مانند metal ions ، cystine) شناسایی کند. به درستی که تنها گونه های دیگر یا گونه های الی بسیار چربی دوست به درون چنین ممبران های نفوذپذیر به گاز توزیع یافته و میگذرند. نوعی از تداخل در نمونه های بالینی به وسیله برخی گازهای بی هوشی مانند nitrous oxide ،halothane ،و isoflurane ایجاد میگردد.این گونه ها همچنین میتوانند از طریق ممبران بیرونی سنسور انتشار یافته ، به طریق الکتروشیمیایی در الکترود پلاتینومی کاهش یابند و در اندازه گیری pO2 سبب مثبت کاذب گردند.با این حال مواد ممبران های نفوذپذیر به گاز که مناسب سازی شده اند و کنترل مناسب پتانسیل بکار گرفته شده در کاتد سنسور به میزان زیادی این مشکل را در دستگاه های نوین کم کرده است.ممبران های بیرونی نفوذپذیر به گاز همچنین انتشار انالیت را به طرف الکترود کار داخلی محدود میکنند و به این ترتیب ممبران میتواند انتقال جرمی انالیت (D/δ در معادله 18( را طوری کنترل کند که در بود و یا نبود جریان همرفتی در نمونه ، انتقال جرمی اکسیژن به سطح الکترود کار پلاتینومی یکسان باشد.
طرح پایه سنسور pO2 کلارک به روش امپرومتری میتواند در شناسایی دیگر گونه های گازی به وسیله تغییر در ولتاژ بکار رفته در الکترود کار استفاده شود.برای نمونه، در شناسایی نیتریک اکساید (NO) با گزینش گری بالا از طرح الکترود گازی مشابه بهره گرفته میشود که در آن پلاتینوم در +0.900 در برابر Ag/AgCl پلاریزه میشود تا NO انتشار یافته را در اند پلاتینومی به نیترات (nitrate) اکسید کند.یک چنین سنسورهای NO را در بررسی های مهم زیست پزشکی در تعیین مقدار NO در نزدیک یا سطح سلولهای تولید کننده NO بکار گرفته اند.
فراتر از دستگاه های امپرومتری، یک روش تخصصی در شناسایی غلظتهای ناچیز از یونهای فلزی سمی در نمونه های بالینی انودیک استریپنیگ ولتامتری (anodi stripping voltammetry = ASV) است.در ASV یک الکترود کار کربنی استفاده میشود (گاهی با یک فیلمی از جیوه بیشتر پوشانده میشود)، و Eappl در آغاز در یک ولتاژ بسیار منفی ثابت میشود، طوری که همه یونهای فلزی در محلول در درون فیلم جیوه و یا روی سطح کربن به فلزهای عنصری کاهش می یابند.سپس Eappl در وضعیت بیشتر مثبت اسکن میشود و فلزهای کاهش یافته رسوب یافته در و یا روی سطح الکترود کار دوباره اکسید میشوند و یک پیک جریانی اندی بزرگی را متناسب با غلظت یونهای فلزی در نمونه اصلی ایجاد میکنند.پتانسیلی که در آن این پیک ها دیده میشوند نشان میدهد که چه فلزی موجود است و بلندای این پیک های جریان بطور مستقیم متناسب با غلظت یون فلزی در نمونه اصلی است.این چنین تکنیک های ASV را میتوان در شناسایی غلظت کلی Pb در نمونه های خون تام بکار گرفت و یک روش غربالگری سریع در مسمویت با سرب و شناسایی میزان در معرض قرارگیری با سرب فراهم کرد.
یک نمونه زیست پزشکی دیگر از ولتامتری مدرن rapid scan cyclic voltammetry است که در اندازه گیری دوپامین در بافت مغز جانوران ازادزی بکار گرفته شده است.در این کاربرد ،اکسیداسیون دوپامین دوپامین به گونه های کینون در یک الکترود میکروکربنی کاشته شده (در ولتاژ نزدیک +0.600 ولت در برابر Ag/AgCl) پیک هایی از جریان ایجاد میکند که متناسب با غلظتهای دوپامین است.از این الکترود میتوان در اندازه گیری این نوروترانسمیتر در بخش های مختلف مغز و یا در یک مکان ثابت استفاده کرد.بیشتر از تحریک های فارماکولوژیک و یا الکتریکی برای اندازه گیری تغییر غلظت محلی دوپامین ناشی از چنین تحریک ها استفاده میشود.
اگر چه تکنیک های ولتامتری / امپرومتری را میتوان در شناسایی بازه گسترده ای از گونه ها استفاده کرد اما گزینشگری این تکنیک ها در نمونه های بالینی پیچیده جایی که بسیاری از گونه های الکترواکتیو دیگر وجود دارند،کمی محدود است.
برای نمونه همانطور که در بحث پیشین گفته شد،در ارتباط با سنسور اکسیژن کلارک ، در غیاب غشا نفوذپذیر به گاز ،دیگر گونه ها که میتوانند در Eappl یکسان یا نزدیک به آن کاهیده شوند مانند اکسیژن میتوانند سب تداخل چشمگیری شوند.
برای گسترش دادن طیف انالیت هایی که با متدهای ولتامتری/امپرومتری شناسایی میشوند، از این تکنیک های الکتروشیمیایی میتوان به عنوان اشکارسازهای بسیار حساس در سیستم های کروماتوگرافی مایع با کارایی بالا استفاده کرد (فصل 13 را ببینید).در کروماتوگرافی مایع با اشکارسازهای الکتروشیمیایی (LC-EC) ،مواد حل شده خارج شونده به وسیله الکترودهایی (معمولا کربن یا جیوه )که جریان از انها میگذرند و حجم مرده بسیار پایینی دارند شناسایی میگردند.(شکل 11-9).

این الکترودها میتوانند به شیوه امپرومتری و یا ولتامتری (با سرعت اسکن بسیار بالا ) عمل کنند، و چندین الکترود میتوانند همزمان بصورت سری یا موازی عمل کنند تا گزینشگری بیشتری بدست اید.برای نمونه،هموسیستئین (homocysteine) را اینگونه میتوان اندازه گیری کرد که 1) افزودن عوامل کاهنده به نمونه سرم تا هموسیستئین ازاد ایجاد شود 2) رسوب دادن پروتیین های نمونه (به کمک تری کلرواستیک اسید) و 3) جداسازی اجزای سرم روی ستون HPLC فاز معکوس اکتادسیل سیلان (reversed-phase octadecylsilane HPLC).هوموسیستئین خارج شونده با روش شناسایی الکتروشیمیایی برخط از طریق اکسیداسیون هوموسیستئین به کمپلکس مرکوریک دی تیولات (mercuric dithiolate complex) مربوط
![]()
و با بکارگیری یک الکترود که از لایه نازکی از مخلوط Hg /Au درست شده است و ولتاژ +0.15 ولت در برابر Ag/AgCl در آن اعمال شده است ،شناسایی و اندازه گیری میشود.یکپارچه کردن باندهای خارج شونده برای هوموسیستئین نتایج کمی با انتخابگری بسیار بالا فراهم میکند.همانند هوموسیستئین ،کتکول ها (catechols) و کتکولامین ها (catecholamines) میتوانند به اسانی و با روش LC-EC مشابه در سرم شناسایی و اندازه گیری شوند به این صورت که کتکول های خارج شونده ،در یک الکترود کاری از جنس کربن با پتانسیل >0.200 v در برابر Ag/AgCl به کینون ها (Quinones) اکسید میشوند.و دیگر اینکه دسته ای از داروهای درمانی نیز به وسیله روش های LC-EC در سرم و یا ادرار شناسایی و اندازه گیری میشوند.
رسانش سنجی (coductometry)
رسانش سنجی تکنیکی الکتروشیمیایی است که برای اندازه گیری یک انالیت موجود در یک مخلوط بکار میرود و با اندازه گیری اثر ان بر رسانش الکتریکی مخلوط تعیین میشود.رسانایی سنجشی از توانایی یونهای محلول است در انتقال جریان تحت اثر یک اختلاف پتانسیل الکتریکی.در یک سلول رسانش سنجی ،پتانسیل الکتریکی بین دو الکترود فلزی بی اثر بکار گرفته میشود.یک پتانسیل متناوب با فرکانس بین 100 تا 3000 هرتز برای پیشگیری از پلاریزاسیون الکترودها بکار میرود.کاهش در مقاومت محلول منجر به افزایش در رسانایی محلول شده و جریان بیشتری بین الکترودها گذر میکند.جریان بدست امده نیز متناوب است.میزان جریان بطور مستقیم متناسب است با رسانایی.رسانایی عکس مقاومت در نظر گرفته میشود و با واحد ohm^-1 (siemens) بیان میشود.در انالیزهای بالینی رسانش سنجی برای اندازه گیری حجم اریتروسیت ها در کل خون (hematocrit) و به عنوان یک مکانیسم انتقال در برخی بیوسنسورها بکار میرود.
اریتروسیت ها به عنوان عایق الکتریکی عمل میکنند چون ترکیب های غشای انها پایه چربی دارند.از این پدیده اول بار در دهه 1940 برای اندازه گیری حجم اریتروسیتها در کل خون (hematocrit)به روش رسانش سنجی بکار برده شد و امروزه برای اندازه گیری هماتوکریت روی دستگاه های چند انالیتی در انالیزهای بالینی استفاده میشود.رسانایی کل خون نه تنها بستگی به حجم و شکل اریتروسیتها دارد بلکه به رسانایی پلاسمای در برگیرنده اریتروسیت ها نیز دارد.افزایش در حجم اریتروسیتها که نسبت به پلاسمای در برگیرنده رسانایی کمتری دارند منجر به کاهش در رسانایی میشود که به وسیله رابطه زیر نشان داده میشود.
![]()
که در ان Gb رسانایی کل خون ، a رسانایی پلاسما ،H درصد هماتوکریت است و c یک فاکتوری برای جهت گیری اریتروسیت ها است.در عمل رسانایی پلاسما دارای فاکتورهای اصلاح کننده برای غلظتهای Na+ و K+ دارد.این کاتیونها معمولا همراه با هماتوکریت روی سیستم های ساخته شده برای انالیزهای بالینی اندازه گیری میشوند.
اندازه گیری هماتوکریت بر پایه رسانایی محدودیتهایی دارد.غلظتهای غیر طبیعی پروتیین رسانایی پلاسما را تغییر میدهد و در اندازه گیری تداخل میکند.غلظتهای کم پروتیین که در رقیق سازی خون با محلول های الکترولیت بدون پروتیین در برخی جراحی ها حاصل میشود سبب گزارش مقدارهای کم و نادرست هماتوکریت به روش رسانش سنجی میشود.متغیرهای پیش از انالیز مانند هم نزدن ناکافی نمونه نیز منجر به نتایج نادرست میشود.هموگلوبین یک انالیت بسیار برتری در پایش خون از دست رفته و نیاز برای انتقال خون در جراحی ها و اسیب ها است.با این حال اندازه گیری های الکتروشیمیایی هماتوکریت همراه با اندازه گیری گازهای خون و دیگر الکترولیتها به دلیل سادگی و اسان بودن روش همچنان بکار گرفته میشود جدای از اینکه محدودیتهایی هم وجود دارد.
یکی دیگر از کاربردهای رسانش سنجی در شمارش الکترونیکی سلول های خونی در یک مخلوط است.طبق اصل کولتر (Coulter principle) رسانایی سلول های خونی نسب به محلول نمکی بکار رفته به عنوان محیط سوسپانسیون کمتر است.سوسپانسیون سلولی را با فشار از یک روزنه بسیار کوچک می گذرانند.دو الکترود در هر یک از دو طرف روزنه جایگذاری شده و یک جریان ثابت بین الکترودها برقرار میشود.هرگاه که یک سلول از روزنه میگذرد ،مقاومت افزایش پیدا میکند و این سبب ایجاد یک قله در اختلاف پتانسیل الکتریکی بین الکترودها میشود.این پالس ها انگاه تقویت شده و شمارش میگردند.
کولومتری (coulometry)
کولومتری بار الکتریکی گذرنده از بین دو الکترود را در یک سلول الکتروشیمیایی اندازه گیری میکند.مقدار بار گذرنده بین الکترودها بطور مستقیم متناسب است با اکسایش یا کاهش یک ماده الکترواکتیو در یکی از الکترودها.شمار کولومب (coulomb) های منتقل شده در این فرایند مرتبط است با مقدار مطلق ماده الکترواکتیو که با قانون فارادی بیان میشود.

![]()
اندازه گیری جریان مرتبط است با مقدار بار گذرنده در واحد زمان (امپر = کولومب بر ثانیه ).کولومتری در کابردهای بالینی در اندازه گیری یون کلراید در سرم یا پلاسما و نیز به عنوان یک روشی از انتقال در برخی بیوسنسورها بکار برده میشود.
تیتراتورهای تجاری شده بر پایه روش کولومتری برای اندازه گیری کلراید طراحی شده اند.یک جریان ثابت بین یک سیم نقره ای (anode) و یک سیم پلاتینیومی (cathode) برقرار میگردد.در اند Ag به Ag+ اکسیده میشود.در کاتد H+ به هیدروژن گازی کاهش پیدا میکند.در یک جریان ثابت بکار رفته شمار کولومب های گذرنده بین اند و کاتد بطور مستقیم متناسب است با زمان (coulombs = amperes * seconds).بنابراین شمار مطلق یونهای نقره تولید شده در اند را میتوان از مقدار زمان جریان گذرنده از آن محاسبه کرد. در حضور یون Cl- ،یونهای Ag+ تشکیل شده به شکل AgCl جامد رسوب میکنند و مقدار یونهای Ag+ ازاد در محلول بسیار کم است.هنگامی که همه یونهای Cl- به شکل کمپلکس در میایند ، یک افزایش ناگهانی در غلظت Ag+ در محلول به وجود میاید.Ag+ های مازاد به روش امپرومتری در یک الکترود دوم از جنس نقره که با یک پتانسیل منفی پلاریزه شده است ،احساس میشوند.Ag+ های مازاد به Ag کاهش پیدا کرده و یک جریان تولید میکنند.هنگامی که این جریان از یک اندازه معین فراتر میرود ،تیتراسیون بازمیایستد.شمار مطلق یونهای Cl- موجود در محلول از میزان زمانی که طی آن تیتراسیون با Ag+ در حال انجام بود محاسبه میشود.با فرض اندازه حجم سرم یا پلاسمایی که در اغاز بکار رفته است امکان محاسبه غلظت یونهای Cl- در نمونه وجود دارد.تیتراسیون به روش کولومتری یکی از صحیح ترین روش های الکتروشیمیایی است چون این روش اندازه مطلق گونه های الکترواکتیو را در نمونه اندازه میگیرد.کولومتری روش استاندارد طلایی در اندازه گیری کلراید در سرم یا پلاسما است. با این حال این روش در معرض مداخله به وسیله انیون های موجود در نمونه با میل ترکیبی بالاتر از کلراید به Ag+ است ، مانند بروماید و به همین دلیل معمولا در ازمایشگاه های بالینی استفاده نمیشود.
سنسورهای نوری شیمیایی (optical chemical sensors)
اوپتود (optode) یک سنسور نوری است که در دستگاه های انالیزی برای اندازه گیری گازهای خونی و الکترولیتها بکار برده میشود.اوپتودها برتری های روشنی نسبت به الکترودها دارند که شامل 1) سادگی ساخت در اندازه های بسیار کوچک 2) نویز بسیار پایین (بی نیاز از سیم های انتقال ) 3) پایداری بسیار زیاد پتانسیل با استفاده از اندازه گیری های نسبت سنجی در چندین طول موج 4) بی نیازی به الکترودهای رفرانس جداگانه است.این برتری ها در ابتدا تکنولوژی سنسورهای نوری را به سوی طراحی سنسورهای گازهای خونی درون رگی تشویق کرد.با این حال همان اصول اشکارسازی پایه ای را میتوان در دستگاهی سازی شیمی بالینی برای نمونه های مختلف بکار برد.در چنین سیستم هایی نور میتواند به مکان اشکارسازی به وسیله رشته های نوری اورده و یا از آن خارج شود و یا بسیار ساده میتوان چشمه های نوری چون دیودهای تابش کننده نور ،فیلترها و اشکارسازهای نوری برای پایش جذب (به وسیله بازتاب ) ،فلورسانس و یا فسفرسانس را جایگذاری کرد(شکل 11-10).

مفاهیم پایه (basic concepts)
سنسورهای نوری برای اندازه گیری PO2 بر پایه نامتحرک سازی برخی رنگهای آلی استوار است رنگهایی مانند pyrene , diphenylphenenthrene , phenenthrene , fluoranthene و یا کمپلکس های لیگاند فلزی (metal ligand complex) مانند ruthenium(II) tris(dipyridine) و متالوپورفیرین های پلاتینیوم (Pt) و پالادیوم (Pd) که درون فیلمهای پلیمری هیدروفوبیک (مانند لاستیک سیلیکونی =silicon rubber) نامتحرک شده اند و اکسیژن درون آنها بسیار محلول است.فلورسانس و یا فسفرسانس چنین گونه هایی در یک طول موج مفروض اغلب در حضور گونه های پارامگنتیک (paramagnetic) و از جمله آنها اکسیژن مولکولی خاموش (quenching) میشوند. در مواردی که رنگهای فلورسنت بکار میرود شدت فلورسانس بازتابی چنین فیلم هایی متناسب با فشار جزیی اکسیژن (PO2) در نمونه که در تماس با فیلم پلیمری قرار میگیرد ، کاهش خواهد یافت، این کاهش در فلورسانس یا میزان quenching مطابق با رابطه stern-volmer است .

که در آن
I0 = شدت فلورسانس در نبود هر گونه اکسیژن است
IPO2 = شدت فلورسانس در یک فشار جزیی مفروض از اکسیژن (PO2) است.
K= ثابت کوانچینگ برای فلوروفور ویژه که بکار رفته است.
بنابراین یک رابطه خطی بین نسبت / IPO2 I0 و PO2 موجود در نمونه وجود دارد.هر چه ثابت استرن – ولمر بزرگتر باشد میزان کوانچینگ برای فلوروفور مفروض نیز بزرگتر است.با این حال مهم است که بدانید ثابت کوانچینگ در یک گستره ای میتواند رفتار استرن – ولمر خطی را در بازه فیزیولوژیک PO2 در خون نشان دهد.برای مثال اگر K خیلی بزرگ باشد انگاه بیشینه کوانچینگ ممکن در گستره ای از PO2 رخ میدهد که کوچکتر از بازه فیزیولوژیک PO2 است.
شدت فسفرسانس یا اندازه گیری طول عمر فسفرسانس کمپلکس های لیگاند فلزی نامتحرک شده هم بکار گرفته میشود ( به عبارت دیگر اتصال اکسیژن طول عمر حالت تحریک شده را کاهش میدهد).سنسورهایی که بر پایه تغییرات در طول عمر لومینسانس هستند این برتری ذاتی را دارند که در برابر اشفتگی طول مسیر نوری و مقدار رنگهای فعال موجود در لایه حسگر (sensing layer) غیر حساس می باشند.
سنسورهای نوری pH نیازمند نامتحرک سازی اندیکاتورهای pH مربوط ( مانند فلوروسئین (fluorescein) ، 8-hydroxy-1,2,6-pyrene trisulfonate (HPTS) ، فنول رد (phenol red) ) درون لایه های نازک پلیمرهای هیدروفیلی (مانند هیدروژل ها = hydrogels) میباشد چون دسترسی تعادلی پروتون ها به اندیکاتور بسیار ضروری است.جذب یا فلورسانس گونه های پروتونه (protonated) یا دپروتونه (deprotonated) میتواند برای اشکار سازی استفاده شود.یک موضوع در رابطه با بکار گیری اندیکاتورهای نامتحرک شده در اندازه گیری صحیح pH فیزیولوژیک ، اثر قدرت یونی روی pKa اندیکاتور است.از انجایی که سنسورهای نوری غلظت رنگهای پروتونه شده یا دپروتونه شده را به عنوان یک سنجش غیر مستقیم از فعالیت یون هیدروژن اندازه گیری میکنند ، تغییرات در قدرت یونی نمونه های فیزیولوژیک میتواند بر صحت اندازه گیری pH اثر بگذارد.
کاربردها (applications)
سنسورهای نوری مناسب برای اندازه گیری pCO2 از مبدل های pH نوری (با اندیکاتورهای نامتحرک شده) به عنوان مبدل های درونی در یک ارایشی مانند طرح سنسور الکتروشیمیایی سبک کلاسیک severinghaus استفاده میکنند(شکل 11-4 را ببینید).افزودن نمک بی کربنات درون لایه هیدروژل حسگر pH یک لایه فیلم مانند الکترولیتی ایجاد میکند که pH آن با بستگی کامل به فشار جزیی دی اکسید کربن (pCO2) در حالت تعادل با فیلم ،تغییر میکند. سنسور pH نوری به وسیله یک فیلم هیدروفوبیک نفوذ پذیر به گاز (مانند لاستیک سیلیکونی)پوشانده میشود تا از دسترسی پروتون جلوگیری کند ولی به CO2 اجازه دهد که با لایه حسگر pH به تعادل برسد.همانطور که فشار جزیی CO2 در نمونه افزایش می یابد، pH لایه بی کربنات کاهش می یابد و کاهش مربوط در شکل دپروتونه اندیکاتور (یا افزایش در شکل پروتونه ) بصورت نوری احساس میشود.
دو شیوه برای حس کردن یونهای الکترولیت به شکل نوری در نمونه های فیزیولوژی بکار رفته است.یک روش ،بسیاری از همان یونوفورهای لیپوفیلیک طراحی شده در ISE های نوع غشا پلیمری (شکل 11-3 را ببینید) را بکار میگیرد.این گونه ها درون فیلمهای پلیمری هیدروفوبی بسیار نازک به همراه یک اندیکاتور pH لیپوفیل درج میگردند.در مورد یونوفورهای کاتیون( برای مثال والینومایسین در حس کردن پتاسیم) ، هنگامی که کاتیون ها به وسیله یونوفور از نمونه به درون فیلم نازک استخراج میشوند،اندیکاتور pH (RH) یک پروتون را به درون فاز نمونه از دست میدهد تا خنثی بودن بار الکتریکی درون فیلم آلی برقرار بماند(R-).این سبب تغییر در جذب نوری یا طیف فلورسانس لایه پلیمری میشود.اگر ضخامت این فیلم ها کمتر از 10 um نگه داشته شود،زمان های پاسخ تعادل به کمتر از 1min خواهد رسید.عمده ترین محدودیت این طراحی این است که pH فاز نمونه هم بر تعادل استخراج سرتاسری یون ها به درون فیلم اثر دارد.بنابراین اندازه گیری همزمان و مستقل pH نمونه، یا رقیق سازی با یک بافر و یا کنترل pH فاز نمونه برای بدست اوردن اندازه گیری های صحیح الکترولیت ها ضروری است.
روش دومی که برای حس کردن یونهای الکترولیت بکار برده میشود نامتحرک سازی یک عامل شناساگر کاتیون و یا انیون در درون یک ماتریکس هیدروژل است درست همانند سنسورهای pH که پیشتر توصیف شدند.عامل شناساگر در این مورد معمولا لیپوفیل نیستند بنابراین باید بصورت کووالانسی در هیدروژل لنگر بیندازند تا بدین وسیله به درون فاز نمونه نشت نکنند. عامل به شیوه ای طراحی شده است که اتصال کاتیون یا انیون انتخابی ،جذب یا طیف فلورسانس گونه های درون هیدروژل را تغییر میدهد.بطور معمول این کار با اتصال ویژگی شناساگری یون و ویژگی کروماتوگرافیک، درون یک مولکول الی انجام میشود. چنین سنسورهای یونی با موفقیت در انالایزرهای گازهای خون و الکترولیت با استفاده از یک ارایه ای از سنسورها با طرح عمومی همانند انچه که در شکل 11-10 نمایش داده شده است، بکار گرفته شده اند.
بیوسنسورها (biosensors)
یک بیوسنسور گونه ویژه ای از سنسور شیمیایی است که از یک بخش شناساگری زیستی و یک بخش مبدل فیزیکوشیمیایی ،اغلب یک وسیله الکتروشیمیایی یا نوری درست شده است. بخش زیستی توانایی شناسایی حضور و فعالیت و یا غلظت یک انالیت خاص را در محلول دارد.شناسایی ممکن است یک واکنش بیوکاتالیزی (بیوسنسورهای بر پایه انزیم=enzyme-based biosensors) باشد و یا یک فرایند اتصالی (بیوسنسورهای بر پایه میل ترکیبی=affinity-based biosensors) هنگامی که بخش شناساگر برای نمونه یک انتی بادی ،بخشی از DNA ،و یا یک گیرنده سلولی باشد.برهم کنش بخش شناساگر با یک انالیت هدف منجر به یک تغییر اندازه گیری شونده در ویژگی محلول ،مانند تشکیل یک محصول یا مصرف یک واکنشگر میشود و این محلول در سطح ابزار جای گرفته است.مبدل تغییر در ویژگی محلول را به یک سیگنال الکتریکی اندازه گیری شونده تبدیل میکند.حالت تبدیل ممکن است یکی از چندین حالت مانند اندازه گیری الکتروشیمیایی یا نوری و اندازه گیری جرم یا گرما باشد.بحث کنونی محدود به بیوسنسورهایی خواهد بود که بر پایه تبدیل الکتروشیمیایی یا نوری کار میکنند چون این گونه بیوسنسورها بخش بزرگی از بیوسنسورهای بکار رفته در کاربری های بالینی را تشکیل میدهند.
بیوسنسورهای پایه انزیمی با اشکارسازی امپرومتری
(enzyme-based biosensors with amperometric detection)
بیوسنسورهای پایه انزیمی با مبدل های الکتروشیمیایی و به ویژه الکترودهای امپرومتری بیشتر از همه در انالیزهای بالینی استفاده شده اند و در نوشتارها به فراوانی از آن ها نام برده شده است. Clark و lyons اول بار بیوسنسورهای امپرومتری را طراحی کردند،آن بیوسنسور گلوکز را درخون اندازه میگرفت و بر پایه نامتحرک سازی گلوکز اکسیداز روی سطح یک سنسور pO2 امپرومتریک طرح شده بود.یک محلول گلوکز اکسیداز به طور فیزیکی میان یک غشا تراوا به گاز الکترود pO2 و یک غشا بیرونی نیمه تراوا به دام می افتد( طرح کلی را در شکل 11-11 ببینید).

غشا بیرونی یک ابزار غربالگری ماده با وزن مولکولی پایین بود تا اجازه دهد سوبسترا (گلوکز) و اکسیژن از غشا بگذرد اما پروتیین ها و دیگر ماکرومولکولها نگذرند.به این شیوه انزیم ها میتوانستند در سطح سنسور تغلیظ شوند.اکسیداسیون گلوکز که به وسیله گلوکز اکسیداز کاتالیز میشود و به صورت زیر است.
![]()
اکسیژن را نزدیک سطح سنسور مصرف میکند.سرعت کاهش در pO2 تابعی است از غلظت گلوکز و به وسیله الکترود pO2 پایش میشود.یک فشار جزیی کاهش یافته و پایدار اکسیژن در یک مدت زمان کوتاه ایجاد میشود و یک مقدار جریان پایدار ازاد میکند که به صورت تابعی از غلظت گلوکز در نمونه کاهش پیدا میکند.
اگر ولتاژ قطبی کننده الکترود pO2 معکوس شود، باعث میشود که الکترود پلاتینیوم نسبت به الکترود رفرانس Ag/AgCl مثبت شود، و اگر غشا تراوا به گاز با یک غشا هیدروفیل که دارای انزیم نامتحرک شده است جایگزین گردد میتوان H2O2 تولید شده به وسیله گلوکز اکسیداز را طبق رابطه زیر اکسید کرد.
![]()
اکنون جریان پایدار ایجاد شده بطور مستقیم متناسب با غلظت گلوکز در نمونه است. در عمل یک ولتاژ به اندازه کافی بالا (اوورپتانسیل = overpotential) باید در پلاتینیوم آند بکار برده شود تا هیدروژن پراکساید را وادار به اکسیداسیون کند.یک ولتاژ خالص +0.7 ولت یا بالاتر از آن (نسبت به Ag/AgCl) معمولا بکار برده میشود.شکل 11-12 طرح پایه شناسایی هیدروژن پراکساید را نمایش میدهد که برای طراحی سنسورهای گلوکز با کاربری بالینی مناسب است اما همچنین برای دیگر سوبستراهایی که انزیم های اکسیداز برای انها هیدروژن پراکساید تولید میکنند نیز مناسب است.

